
")

Четкое и ясное представление об этом дал Дмитрий Рождественский, заместитель начальника отдела координации формирования общих рынков лекарственных средств и медицинских изделий Департамента технического регулирования и аккредитации ЕАЭК. Его доклад, озвученный на Фармацевтическом Форуме стран СНГ и ЕАЭС, стал логическим продолжением выступления Дмитрия Щекина. Дмитрий Рождественский пояснил, что до января 2021 года в странах ЕАЭС, наряду с едиными процедурами регистрации ЛС и МИ общего рынка ЕАЭС, сохранятся национальные процедуры (рисунок). Таким образом, на территориях стран ЕАЭС некоторое время параллельно будут существовать две системы регистрации. Этот переходный период был определен по настоянию фармацевтического бизнеса, так как в ходе обсуждений, выяснилось, что компании не были готовы предоставлять досье по единым правилам и предложили более комфортную для себя схему.
Рисунок
Рынок ЛС в рамках реализации Соглашения
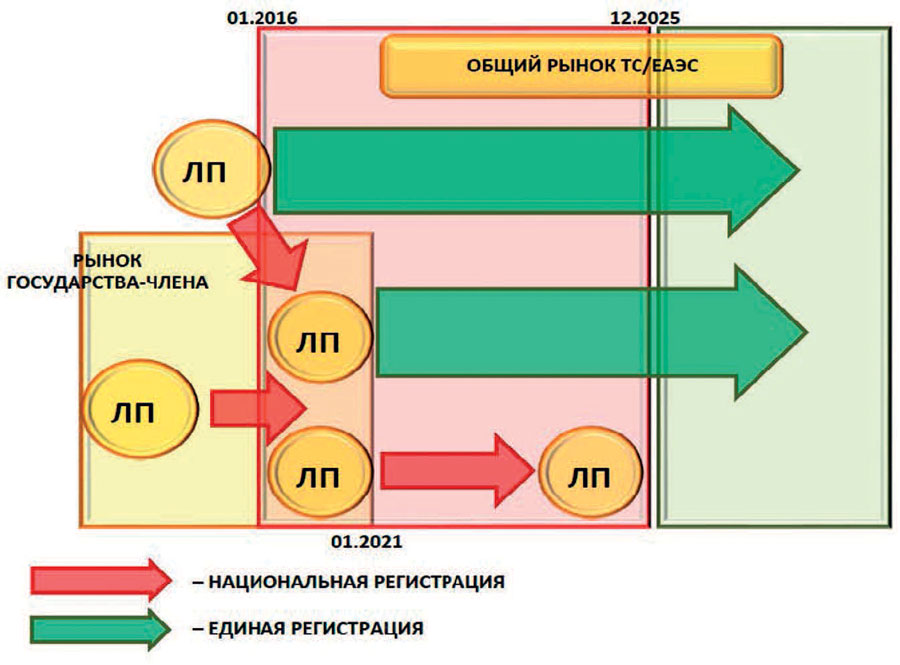
В целом в течение переходного периода в ЕАЭС предусмотрено 5 разновидностей процедур регистрации ЛС. Первая и вторая - это единая регистрация ЛС. Она, как уже говорилось выше, станет возможной после подписания сторонами полного пакета документов и установления тарифов на регистрацию национальными регуляторными органами (по словам выступающих, на это потребуется минимум полгода). Единая регистрация будет выполняться по принципам децентрализованной процедуры и процедуры взаимного признания. Поскольку не все понимают, как будет работать единый рынок лекарств даже в рамках этих двух процедур регистрации, было решено не вводить в схему централизованную процедуру регистрации, так как она требует создания наднационального регуляторного органа (по словам участников форума, - это дело будущего).
Третьей процедурой является приведение досье в соответствие с правилами ЕАЭС. Она предназначена для препаратов, которые зарегистрированы/будут зарегистрированы по национальным правилам, как по состоянию на 2016 год, так и в течение периода с 2016 по 2021 годы. После 31 декабря 2025 года она естественным образом «отомрет» и ее больше не станет, соответственно, не будет и тех лекарственных средств, досье которых к этому сроку не будут приведены в соответствие с правилами ЕАЭС. Длительность процедуры приведения досье в соответствие с правилами ЕАЭС составляет 100 дней. Важным вопросом является выбор референтного государства. Если ЛС зарегистрировано в одном государстве-члене ЕАЭС, то референтным может только данное государство. При регистрации ЛС в нескольких государствах-членах ЕАЭС, в качестве референтного может выбрано любое из этих государств. Досье представляется в формате CTD. Приведение досье в соответствие будет включать в себя изготовление модулей 1, 2 и 3. Модули 4 и 5 комплектуются имеющимися доклиническими и клиническими исследованиями без их приведения в соответствие с требованиями к оформлению отчетной документации при условии, что они проведены в соответствии с требованиями GLP и GCP.
Четвертая процедура представляет собой подтверждение регистрации/перерегистрации. Она предназначена для тех лекарственных средств, которые прошли процедуру единой регистрации. Первое регистрационное удостоверение у ЛС, зарегистрированных по единой процедуре, будет выдаваться на 5 лет. После подтверждения регистрации (через 5 лет) на них будет выдаваться бессрочное регистрационное удостоверение.
При этом будут два исключения. Первое - будет касаться препаратов, которые зарегистрированы по национальной процедуре на территории как минимум трех государств-членов ЕАЭС в течение как минимум 5 лет. Эти препараты после приведения своего досье в соответствие с правилами ЕАЭС будут получать сразу бессрочное удостоверение.
Второе - будет применяться к препаратам, имеющим особенности регистрационного досье, которые не позволят регуляторному органу однозначно определить его эффективность и безопасность. К таким особенностям относятся ЛС, для которых необходимо:
- принятие дополнительных мер для обеспечения безопасного применения ЛС;
- проведение пострегистрационных исследований безопасности ЛС;
- установление дополнительных требований к регистрации ЛС и подаче сообщений о подозреваемых нежелательных реакциях;
- принятие других условий или ограничений в целях безопасного и эффективного применения ЛС в соответствии с требованиями GVP;
- проведение пострегистрационных исследований эффективности ЛС, для установления аспектов эффективности, которые не могут быть исследованы до его реализации;
- проведение пострегистрационных исследований безопасности в случае наличия опасений по поводу рисков данного ЛС;
- проведение пострегистрационных исследований эффективности, если понимание заболевания или клиническая методология показывают, что предыдущие оценки эффективности требуют существенного пересмотра.
В этих случаях регистрационное удостоверение выдается на 5 лет, с последующим продлением на 5 лет и лишь затем бессрочно, либо ежегодно проводится переоценка соотношения польза/риск и при признании его неблагоприятным регистрационное удостоверение отменяется (аннулируется).
И, наконец, пятая разновидность - регистрация по национальным правилам. С января 2021 года она прекратит свое существование. Небольшие производители, которые хотят работать только на внутреннем рынке своей страны, в рамках единой процедуры взаимного признания должны будут указать только одну страну. ЛС, которые были зарегистрированы по национальной процедуре до 2021 года, как в течение переходного периода, так и до него, будут продолжать свое обращение до декабря 2025 года. Если их производители не приведут свое досье в соответствие с едиными правилами, то в декабре 2025 года обращение таких ЛС прекратиться, вне зависимости от того, какое регистрационное удостоверение (срочное или бессрочное) они имели. Т.е. даже в случае наличия бессрочного удостоверения их обращение будет остановлено. Поэтому с 2016 по 2025 годы производители таких лекарственных средств должны привести досье в соответствие с едиными правилами.
Сроки осуществления единой процедуры регистрации регламентированы и составляют 210 дней. Движение досье по этапам расписано буквально по дням. Но есть интересные особенности, которые необходимо обязательно учитывать. Первая - сроки до 14 дней, указанные в правилах, подразумевают рабочие дни, все сроки больше 14 дней в правилах приведены в календарных днях. Связаны эти особенности с тем, что в наших государствах дается разное по продолжительности время отдыха на государственные праздники. Вторая особенность - время, предоставляемое фармацевтической компании для ответов на вопросы экспертной организации. В рамках процедуры регистрации предусмотрен, так называемый, стоп-тайм продолжительностью в 90 или 180 дней. Т.е. выполнение процедуры регистрации и перерегистрации на время, которое берет заявитель для предоставления ответов на вопросы экспертов, останавливается. Максимальная продолжительность стоп-тайма, как видно на рисунке, составляет 180 дней. Заявитель, по своему выбору, может взять как все 180 дней сразу, так и разделить этот срок на части, но при этом следует помнить, что дополнительных сроков приостановления процедуры регистрации предоставляться не будет. Если весь срок стоп-тайма исчерпан, то заявителю придется отвечать на вопросы экспертов срочно в режиме онлайн.
Процедура взаимного признания выполняется в виде последовательной оценки. Сначала регистрационное досье проходит экспертизу в референтном государстве. По ее результатам заявитель получает регистрационное удостоверение и экспертный отчет, который он потом подает в государство признания. Сделать это заявитель может через 1, 2 или 3 года, но выдача регистрационного удостоверения в государстве признания будет привязана к дате референтного государства. Это сделано для удобства заявителей, чтобы все сроки при последующей перерегистрации подходили синхронизировано.
Начальные этапы децентрализованной процедуры регистрации и процедуры взаимного признания схожи. Но затем к работе над досье подключаются все государства признания, которые укажет заявитель. При этом референтное государство составляет промежуточный отчет, которые государства признания оценивают и имеют при этом право дополнить своими замечаниями. Заявитель дает ответы на эти замечания и затем одномоментно всеми участвующими в процедуре государствами принимается решение о регистрации или отказе в регистрации.
Обязательным компонентом досье является сертификат GMP ЕАЭС. Однако, появятся такие сертификаты только после начала функционирования единого рынка лекарств и на начальном этапе у всего фармацевтического бизнеса их просто не будет. Поэтому в правилах регистрации предусмотрено, что при отсутствии сертификата GMP ЕАЭС заявитель может представить национальный сертификат GMP (на производственную площадку, выпускающую готовую лекарственную форму или осуществляющую выпускающий контроль качества), выданный уполномоченным органом государства-члена ЕАЭС, или отчет об инспектировании. Высочайшим проявлением здравого смысла является то, что все государства-члены ЕАЭС согласились признавать сертификаты друг друга. Причем референтная страна признает сертификат соответствия требованиям GMP, выданный любой из стран ЕАЭС.
При отсутствии национального сертификата GMP государств-членов ЕАЭС выполняется внеплановое обязательное инспектирование в рамках регистрации без продления ее срока. Помимо этого, к критериям назначения GMP инспекции относятся: отсутствие сертификата GMP ЕАЭС или регистрации ЛС в государстве-члене ЕАЭС, произведенных на данной производственной площадке, а также выявление в ходе экспертизы регистрационного досье фактов, ставящих под сомнение достоверность сведений.
Клинические исследования (далее - КИ), представленные в досье, рассматриваются в процессе экспертизы при соблюдении ОДНОГО из следующих условий:
- КИ проведены до 1 января 2016 года или продолжаются по состоянию на эту дату (набор пациентов не закрыт) на территории ЕАЭС;
- КИ проведены до 1 января 2016 года на территории государств-членов ICH и зарегистрированы по результатам КИ в государствах-членах ICH;
- КИ, инициированные после 1 января 2016 года, должны быть полностью или частично (в части, связанной с набором пациентов) проведены в клиниках на территории стран ЕАЭС.
Если перечисленные условия не выполняются, заявитель обязан провести/повторить КИ (как минимум одно) на территории стран ЕАЭС или по решению уполномоченного органа ему будет назначена инспекция клинической базы.
Требования к формированию регистрационного досье изложены в пяти приложениях к правилам регистрации и экспертизы. Приложение №1 содержит требования к составу досье в целом и для различных групп лекарственных препаратов; Приложение №2 - формы заявлений для подачи досье на регистрацию, подтверждение регистрации и внесение изменений; Приложение №3 - требования к оформлению нормативного документа по контролю качества; Приложение №4 - краткий перечень документов регистрационного досье; Приложение №5 - указания по принципам размещения документов в разделах модулей досье. Особое внимание участников конференции докладчик обратил на Приложение №3. Учитывая, что единый рынок начинает работать в условиях, когда Фармакопея ЕАЭС только начинает разрабатываться, стороны пришли к соглашению, что нормативный документ по контролю качества заявители могут разрабатывать по требованиям референтного государства. И этот документ будет приниматься государствами признания.
***
Следует отметить, что условия, при которых результаты КИ принимаются на рассмотрение при регистрации, вызвали многочисленные споры у участников конференции. Особенно этот вопрос волновал отечественных производителей, которые готовились к началу работы единого фармацевтического рынка и заранее проводили клинические исследования. Из-за отсрочки все их усилия и вложенные средства пропадают даром, а проведенные исследования, если не будут сдвинуты сроки, не будут признаны. Ответ на этот вопрос не был получен, и он так и остался открытым.
На форуме неоднократно отмечалось, что все же, в будущем, не исключено создание наднационального органа по регистрации и экспертизе. Но это вопрос политической воли и как будет на самом деле покажет время.
Ольга Баимбетова.








