
")

В соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года, статьями 4 и 13 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года Коллегия Евразийской экономической комиссии решила:
1. Утвердить прилагаемое Руководство по составлению нормативного документа по качеству лекарственного препарата.
2. Настоящее Решение вступает в силу по истечении 6 месяцев с даты его официального опубликования.
Председатель Коллегии Евразийской экономической комиссии Т. Саркисян
УТВЕРЖДЕНО
Решением Коллегии
Евразийской экономической комиссии
от 7 сентября 2018 г. № 151
РУКОВОДСТВО
по составлению нормативного документа по качеству лекарственного препарата
I. Общие положения
1. Настоящее Руководство разработано с учетом приложения № 3 к Правилам регистрации и экспертизы лекарственных средств для медицинского применения, утвержденным Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 78 (далее – Правила) и устанавливает порядок составления нормативного документа по качеству лекарственного препарата (далее – нормативный документ по качеству).
2. Настоящее Руководство распространяется на лекарственные препараты независимо от их происхождения (химические, биологические, растительные и т. п.).
3. Нормативный документ по качеству устанавливает требования к контролю качества лекарственного препарата (содержит спецификацию и описание методик испытаний или ссылки на них, а также соответствующие критерии приемлемости показателей качества и т. п.) на основании проведенной экспертизы лекарственного препарата, утверждается уполномоченным органом государства – члена Евразийского экономического союза (далее соответственно – государство-член, Союз) при регистрации лекарственного препарата и предназначен для контроля качества лекарственного препарата в пострегистрационный период на территориях государств-членов.
4. Нормативный документ по качеству составляется исключительно в отношении лекарственного препарата.
В отношении активной фармацевтической субстанции составление нормативного документа по качеству не требуется.
5. Нормативный документ по качеству содержит сведения о качестве лекарственного препарата, включенные в разделы 3.2.P.1, 3.2.P.5.1, 3.2.P.5.2, 3.2.P.7 и 3.2.P.8.1 модуля 3 регистрационного досье лекарственного препарата (приложение № 4 к Правилам) и используемые контрольными лабораториями государств-членов,
не имеющими доступа к модулю 3, для осуществления контроля качества лекарственных препаратов. Сведения, содержащиеся в
модуле 3 регистрационного досье лекарственного препарата, имеют первостепенное значение. Сведения, содержащиеся в нормативном документе по качеству, не могут противоречить сведениям, содержащимся в модуле 3 регистрационного досье лекарственного препарата.
6. Нормативный документ по качеству составляется с учетом указаний, предусмотренных приложением № 3 к Правилам.
7. Спецификации на активные фармацевтические субстанции, полученные путем химического синтеза, и содержащие их лекарственные препараты составляются в соответствии с требованиями согласно приложению № 1.
8. Настоящее Руководство применяется в отношении следующих лекарственных форм:
твердые лекарственные формы для приема внутрь;
жидкие лекарственные формы для приема внутрь;
парентеральные лекарственные формы (большого и малого объемов).
Порядок составления нормативного документа по качеству и спецификаций в отношении указанных лекарственных форм может применяться для иных лекарственных форм.
9. Общие требования к составлению спецификаций на лекарственные препараты и активные фармацевтические субстанции биологического происхождения установлены в главе 6 Правил проведения исследований биологических лекарственных средств Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 89.
10. Общие требования к составлению спецификаций на лекарственные растительные препараты устанавливаются в руководстве по выбору тестов и критериев приемлемости для составления спецификаций на препараты из лекарственного растительного сырья, утверждаемом Евразийской экономической комиссией.
11. Требования к составлению спецификаций на лекарственные препараты отдельных видов и активные фармацевтические субстанции, входящие в их состав, в зависимости от их лекарственной формы или свойств действующего вещества определяются соответствующими актами, входящими в право Союза.
12. В отношении лекарственных препаратов, указанных в
пунктах 8 – 10 настоящего Руководства, а также радиофармацевтических препаратов настоящее Руководство содержит исключительно требования к оформлению нормативного документа по качеству.
II. Структура нормативного документа по качеству
13. Структура нормативного документа по качеству должна соответствовать приложению № 3 к Правилам и содержать 8 разделов в следующем порядке:
а) титульный лист по форме согласно приложению № 2;
б) состав лекарственного препарата;
в) спецификация;
г) описание методик испытаний;
д) описание упаковки;
е) маркировка;
ж) условия хранения;
з) срок годности (хранения).
14. Показатели качества и регламентируемые нормы приводятся согласно спецификации производителя на конец срока годности (хранения). При наличии одного и того же показателя качества в спецификациях на выпуск и на конец срока годности (хранения) регламентируемые нормы для такого показателя приводятся в нормативном документе по качеству согласно спецификации производителя на конец срока годности (хранения).
1. Титульный лист
15. В форме титульного листа нормативного документа по качеству указываются:
а) все торговые наименования лекарственного препарата, согласованные уполномоченным органом государства-члена и включенные в регистрационное удостоверение, и лекарственная форма в соответствии с Номенклатурой лекарственных форм, утвержденной Решением Коллегии Евразийской экономической комиссии
от 22 декабря 2015 г. № 172;
б) дозировка в соответствии с принципами, указанными в требованиях к инструкции по медицинскому применению лекарственных препаратов и общей характеристике лекарственных препаратов для медицинского применения, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 88 (далее – требования к инструкции);
в) полное и (или) сокращенное наименования и страна держателя регистрационного удостоверения.
16. Необходимо предусмотреть специальные поля для указания номера нормативного документа по качеству и грифа согласования. Требования к указанию номера нормативного документа по качеству приводятся в приложении № 3 к Правилам. В нормативном документе по качеству не допускается перечислять участников процесса производства лекарственного препарата.
2. Состав лекарственного препарата
17. Состав лекарственного препарата приводится в соответствии с разделом 3.2.Р.1 модуля 3 регистрационного досье лекарственного препарата (без указания функционального назначения вспомогательных веществ) в отдельном разделе нормативного документа по качеству путем указания качественного и количественного состава активных фармацевтических субстанций и вспомогательных веществ (с указанием ссылок на Фармакопею Союза, а при отсутствии в ней – на фармакопеи государств-членов или на нормативные документы, регламентирующие их качество).
18. Порядок изложения раздела 3.2.Р.1 модуля 3 регистрационного досье лекарственного препарата приведен в приложении № 1 к Правилам.
3. Спецификация
19. Спецификация должна представлять собой копию документа, содержащегося в разделе 3.2.Р.5.1 модуля 3 регистрационного досье лекарственного препарата. Спецификация представляется в виде таблицы, состоящей из 3 граф:
а) показатели качества;
б) нормы (допустимые пределы);
в) ссылки на методы испытаний.
20. Показатели качества устанавливаются в соответствии с требованиями общих фармакопейных статей (монографий) Фармакопеи Союза, а при отсутствии в ней – в соответствии с требованиями общих фармакопейных статей (монографий) фармакопей государств-членов, с учетом особенностей конкретной лекарственной формы лекарственного препарата в зависимости от физико-химических (биологических) свойств активной фармацевтической субстанции и в соответствии с настоящим Руководством.
21. Наименования показателей качества в спецификации указываются в соответствии с Фармакопеей Союза, а при отсутствии в ней – в соответствии с фармакопеей референтного государства.
4. Описание методик испытаний
22. Описание методик испытаний лекарственного препарата по всем показателям качества, указанным в спецификации, со ссылками на Фармакопею Союза, а при отсутствии в ней – на фармакопеи государств-членов, приводится в соответствии с разделом 3.2.Р.5.2 модуля 3 регистрационного досье лекарственного препарата.
5. Описание упаковки
23. В разделе 5 нормативного документа по качеству необходимо описать:
а) первичную упаковку (ампулы, флаконы, банки, пакеты и т. п.);
б) количество единиц продукции в первичной упаковке (например, количество таблеток в контурной ячейковой или безъячейковой упаковке);
в) промежуточную, вторичную (потребительскую) упаковку и количество первичных упаковок в ней (например, количество контурных ячейковых упаковок во вторичной упаковке);
г) наличие поглотителя влаги, листка-вкладыша (инструкции по медицинскому применению), комплектность (игла, капельница, зажим и т. п.) и другие сведения в соответствии с разделом 3.2.P.1 модуля 3 регистрационного досье лекарственного препарата. Кроме того, требования к описанию характера и содержимого упаковки установлены в разделе 6.5 требований к инструкции. Характеристику упаковки приводить не требуется, однако, если такие сведения являются обязательными, они не должны противоречить разделу 3.2.P.7 модуля 3 регистрационного досье лекарственного препарата.
6. Маркировка
24. В разделе 6 нормативного документа по качеству необходимо указать ссылку на раздел 1.3.2 модуля 1 регистрационного досье лекарственного препарата.
7. Условия хранения
25. Сведения об условиях хранения не должны противоречить сведениям, содержащимся в разделе 3.2.P.8 модуля 3 регистрационного досье лекарственного препарата.
26. Общие требования к описанию условий хранения приводятся в приложении № 6 к требованиям к инструкции.
8. Срок годности (хранения)
27. Сведения, включаемые в раздел 8 нормативного документа по качеству, не должны противоречить сведениям, содержащимся в разделе 3.2.P.8 модуля 3 регистрационного досье лекарственного препарата. Общие требования к указанию сроков годности (хранения) приведены в разделе 6.3 требований к инструкции.
III. Оформление нормативного документа по качеству
28. Текст нормативного документа по качеству должен быть кратким, без повторов и исключать возможность двоякого толкования. Сокращение слов в тексте, наименованиях рисунков и схем не допускается, исключение составляют сокращения, содержащиеся в спецификации и установленные Фармакопеей Союза, а при отсутствии в ней – фармакопеями государств-членов.
29. Требования к качеству лекарственного препарата излагаются в повелительной форме, а методики испытаний – в форме третьего лица множественного числа.
30. Если методика испытания, требования к показателям качества, их нормы и отклонения от них, указанные в нормативном документе по качеству, установлены Фармакопеей Союза, а при отсутствии в ней – в фармакопеях государств-членов, следует указывать ссылку на источник без описания методики испытания. При указании требований и показателей качества, установленных фармакопеями третьих государств, следует представлять описание используемых методик испытания со ссылкой на источник.
31. Термины, обозначения и определения должны соответствовать Фармакопее Союза, а при отсутствии в ней – фармакопеям государств-членов. При использовании терминов и обозначений, которые не установлены актами органов Союза в сфере обращения лекарственных средств (в том числе Фармакопеей Союза) или фармакопеями государств-членов и не являются общепризнанными, следует в тексте приводить их определения.
32. В тексте не допускается:
а) применение оборотов разговорной речи;
б) применение для одного и того же понятия различных терминов, близких по смыслу (синонимов), а также иностранных слов и терминов при наличии равнозначных слов и терминов в русском языке;
в) сокращение обозначений единиц измерения, если они употребляются без цифр;
г) замена слов буквенными обозначениями (за исключением таблиц и формул);
д) применение математических знаков без цифр.
33. При описании аналитических методик в отношении применяемых реактивов, стандартных растворов, буферных растворов и материалов необходимо указать обозначения стандартов или технические условия, а также полное и (или) сокращенное наименования юридического лица – производителя. При наличии в Фармакопее Союза или в фармакопеях государств-членов описания применяемых при испытаниях реактивов, стандартных растворов, буферных растворов и материалов их наименования выделяются курсивом и после них указывается обозначение «Р». При отсутствии в Фармакопее Союза или в фармакопеях государств-членов описания применяемых реактивов, стандартных растворов, буферных растворов и материалов необходимо указать обозначения стандартов или регламентирующие их технические условия, а также наименование юридического лица – производителя. Курсивом выделяются также наименования титрованных растворов, описанных в Фармакопее Союза или в фармакопеях государств-членов, без указания обозначения «Р». Для применяемой мерной посуды указывается ее вместимость.
34. Формулы расчета должны быть представлены в развернутой и сокращенной формах и сопровождаться пояснением указанных в них физических величин. Обозначения физических величин должны быть приведены в соответствии с требованиями Фармакопеи Союза, а при отсутствии в ней – в соответствии с требованиями фармакопей государств-членов. Перенос части формулы расчета на другую строку не допускается.
35. Для измерения физических величин, указанных в нормативном документе по качеству, применяются единицы измерения, предусмотренные Международной системой единиц (СИ), и единицы измерения, используемые наравне с ними.
36. Текст нормативного документа по качеству оформляется с учетом следующих параметров настройки:
размеры полей: левое – 30 мм, правое – 15 мм, верхнее и нижнее – 20 мм;
абзацный отступ – 12,5 мм;
шрифт Times New Roman размера № 14 (для номера нормативного документа по качеству – 16).
Заголовки и наименование лекарственного препарата начинаются с прописной буквы и выделяются полужирным шрифтом.
Основной текст печатается через 1,5 междустрочного интервала, текст в спецификации и примечаниях – через 1 междустрочный интервал, текст в заголовках и в описании качественного и количественного состава – через 1 междустрочный интервал (в случае указания разных наименований – через 1,5 междустрочного интервала).
37. Страницы нормативного документа по качеству должны быть пронумерованы. При этом на первой странице номер не проставляется.
38. Рисунки, схемы, диаграммы, графики, спектры и хроматограммы могут быть выполнены на отдельных страницах или в тексте нормативного документа по качеству.
____________
ПРИЛОЖЕНИЕ № 1
к Руководству
по составлению нормативного документа по качеству лекарственного препарата
ТРЕБОВАНИЯ
к составлению спецификаций: аналитических методик
и критериев приемлемости фармацевтических субстанций, полученных путем химического синтеза, и содержащих их лекарственных препаратов
I. Общая характеристика
1. Настоящие Требования содержат описание подходов к разработке единого комплекса спецификаций на активные фармацевтические субстанции (действующие вещества) и (или) лекарственные препараты, в том числе рекомендации по разработке и обоснованию критериев приемлемости (допустимых норм) и выбору аналитических методик для активных фармацевтических субстанций, полученных путем химического синтеза, и содержащих их лекарственных препаратов.
2. Спецификация задает совокупность критериев, которым должны соответствовать активные фармацевтические субстанции и (или) лекарственные препараты, чтобы считаться пригодными для своего целевого назначения. Соответствие спецификации означает, что активная фармацевтическая субстанция и (или) лекарственный препарат соответствуют приведенным критериям приемлемости при условии, что испытания проведены согласно аналитическим методикам, указанным в ней. Спецификации являются ключевыми стандартами качества, которые предлагает и обосновывает производитель и утверждает уполномоченный орган государства – члена Евразийского экономического союза (далее соответственно – государства-члены, Союз) при регистрации лекарственного препарата.
Спецификации являются одним из элементов общей стратегии контроля активных фармацевтических субстанций и (или) лекарственных препаратов, разработанной с целью обеспечения их качества и постоянства характеристик. Другие элементы этой стратегии включают доскональное установление всех характеристик активных фармацевтических субстанций, промежуточных продуктов и (или) лекарственных препаратов (далее – продукты) в процессе разработки лекарственного препарата, а также строгое соблюдение Правил надлежащей производственной практики Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 77 (далее – Правила надлежащей производственной практики), например, подходящие помещения и оборудование, валидированный процесс производства лекарственного препарата, валидированные аналитические методики, испытание исходных материалов и сырья, внутрипроизводственные испытания, испытание стабильности и т. д.
Спецификации предназначены для подтверждения качества активной фармацевтической субстанции и лекарственного препарата. Они не преследуют цели полного установления характеристик и поэтому должны быть основаны на характеристиках, подтвердивших свою пригодность для обеспечения безопасности и эффективности активной фармацевтической субстанции и лекарственного препарата.
II. Сфера применения
3. Качество активных фармацевтических субстанций и (или) лекарственных препаратов определяется уровнем разработки, внутрипроизводственным контролем, контролем соблюдения Правил надлежащей производственной практики, валидацией процесса производства, а также спецификациями, применяемыми к ним в процессе разработки и производства. В настоящих Требованиях определяются требования к спецификации, т. е. к тем испытаниям, методикам и критериям приемлемости, которые обеспечивают качество активной фармацевтической субстанции и лекарственного препарата на момент выпуска и в течение всего срока годности (хранения). Спецификации являются важным, но не единственным компонентом обеспечения качества активных фармацевтических субстанций и лекарственных препаратов.
4. Настоящие Требования применяются в отношении лекарственных препаратов (включая входящие в их состав активные фармацевтические субстанции), находящихся на этапе регистрации.
5. В настоящих Требованиях не рассматриваются активные фармацевтические субстанции и (или) лекарственные препараты, находящиеся на этапе клинической разработки.
Настоящие Требования могут применяться в отношении синтетических и полусинтетических антибиотиков и низкомолекулярных синтетических пептидов, однако положения настоящих Требований недостаточны для надлежащего описания спецификаций высокомолекулярных пептидов и полипептидов, а также биотехнологических (биологических) препаратов.
Радиофармацевтические препараты, продукты ферментации, олигонуклеотиды, растительные препараты и необработанные препараты животного и растительного происхождения в настоящих Требованиях не рассматриваются.
6. В настоящих Требованиях приводятся рекомендации в отношении критериев приемлемости, которые необходимо разработать для всех активных фармацевтических субстанций и (или) лекарственных препаратов, т. е. универсальных критериев приемлемости, а также специальных критериев приемлемости, предусмотренных для отдельных активных фармацевтических субстанций и (или) лекарственных препаратов. Настоящие Требования следует рассматривать как основное руководство по составлению спецификаций и выбору критериев приемлемости. При этом при появлении новых аналитических технологий и модификаций существующих технологий при достаточном обосновании следует также использовать их данные.
7. В настоящих Требованиях рассматриваются следующие лекарственные формы:
а) твердые лекарственные формы для приема внутрь;
б) жидкие лекарственные формы для приема внутрь;
в) парентеральные лекарственные формы (большого и малого объемов).
8. Указанные в пункте 6 настоящих Требований лекарственные формы служат моделями, которые могут быть применимы к иным лекарственным формам. При составлении спецификаций на другие лекарственные формы (например, ингаляционные (порошки, растворы и т. д.), лекарственные формы для местного применения (кремы, мази, гели) и трансдермальные лекарственные формы) целесообразно прибегать к расширению концепций, используемых в настоящих Требованиях.
8. В настоящих Требованиях представлено краткое описание каждой концепции и указаны обстоятельства, при которых они могут быть применимы. Как правило, предложения по применению этих концепций должны быть обоснованы заявителем и перед выпуском согласованы с уполномоченным органом государства-члена.
III. Определения
9. Для целей настоящих Требований используются понятия, которые означают следующее:
«быстрорастворяющиеся лекарственные препараты» – твердый лекарственный препарат для приема внутрь с немедленным высвобождением считается быстрорастворяющимся, если не менее 80 % от заявленного содержания фармацевтической субстанции растворяется в течение 15 минут в каждой из следующих сред: при pH 1,2, 4,0 и 6,8;
«внутрипроизводственные испытания» – испытания, проводимые в процессе производства фармацевтической субстанции и (или) лекарственного препарата, а не в рамках комплекса испытаний, проводимых перед выпуском в обращение;
«комбинированный препарат» – лекарственный препарат, содержащий более одной фармацевтической субстанции;
«немедленное высвобождение» – процесс растворения лекарственного препарата в желудочно-кишечном содержимом
без намерения отсрочки или пролонгации его растворения или абсорбции;
«отложенное (отсроченное) высвобождение» – высвобождение фармацевтической субстанции в момент времени, не совпадающий со временем после непосредственного приема внутрь;
«полиморфизм» – нахождение одной и той же фармацевтической субстанции в различных кристаллических формах, в том числе в виде продуктов сольватации или гидратации (псевдополиморфы) и аморфных форм;
«продукт деградации (продукт разложения)» – молекула, образующаяся вследствие химического изменения молекулы фармацевтической субстанции со временем и (или) под воздействием освещения, температуры, pH, влаги и т.д. или при взаимодействии
с вспомогательным веществом и (или) системой упаковки (укупорки);
«пролонгированное высвобождение» – процесс высвобождения фармацевтической субстанции на протяжении длительного периода после введения;
«растворитель» – неорганическая или органическая жидкость, используемая в качестве среды (носителя) для приготовления растворов и суспензий при синтезе фармацевтической субстанции или производстве лекарственного препарата;
«рацемат» – смесь (твердая, жидкая, газообразная) или раствор эквимолярных количеств двух энантиомеров. Не обладает оптической активностью;
«специфичное испытание» – испытание, считающееся применимым в отношении определенных фармацевтических субстанций и (или) определенных лекарственных препаратов в зависимости от их особенностей и (или) целевого назначения;
«стандартный образец (стандартный материал)» – вещество, используемое в качестве стандарта (эталона) при количественном определении, идентификации или испытании на чистоту;
«универсальное испытание» – испытание, считающееся потенциально применимым ко всем фармацевтическим субстанциям
и (или) ко всем лекарственным препаратам (например, испытания при описании, идентификации, количественном определении и определении примесей);
«фармацевтические субстанции, хорошо растворимые в воде» – фармацевтические субстанции с соотношением «доза вещества / растворимость вещества» меньшим или равным 250 мл в диапазоне pH 1,2 – 6,8 (например, соединение А обладает наименьшей растворимостью, равной 1 мг/мл при 37 ± 0,5 ºС и pH 6,8, представлено в трех дозировках: 100, 200 и 400 мг. Такой лекарственный
препарат считается малорастворимым, поскольку его соотношение «доза вещества / растворимость вещества» составляет 400 мл
(400 мг : 1 мг/мл = 400 мл) и тем самым превышает 250 мл);
«хиральный» – не совпадающий при наложении зеркального отражения, применяется в отношении молекул, конформаций и макроскопических объектов (например, кристаллов). Термин также распространяется на образцы веществ, чьи молекулы хиральны,
даже если макроскопическое множество молекул представляет собой рацемат;
«энантиомеры» – изомеры, которые имеют одинаковые состав и химическое строение, но отличаются пространственным расположением атомов в молекуле и являются несовместимыми зеркальными отражениями.
Для целей настоящих Требований понятие «примесь» применяется в значении, определенном в Правилах надлежащей производственной практики, понятие «критерии приемлемости» применяется в значении, определенном в Руководстве по качеству лекарственных растительных препаратов (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 10 мая 2018 г. № 6), понятие «модифицированное высвобождение» применяется в значении, определенном в Руководстве по качеству лекарственных препаратов с модифицированным высвобождением для приема внутрь (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 16 февраля 2018 г. № 2).
IV. Виды испытаний
1. Периодические (выборочные) испытания
10. Периодические (выборочные) испытания – проведение определенных испытаний при выпуске в обращение на заранее выбранных сериях активной фармацевтической субстанции и (или) лекарственных препаратов (далее – серии) и (или) через заранее установленные промежутки времени (а не относительно каждой серии). При этом серии, не подвергаемые испытаниям, также должны соответствовать всем критериям приемлемости, предусмотренным для активной фармацевтической субстанции и (или) лекарственных препаратов. Проведение такого испытания представляет собой неполную программу испытаний, и, следовательно, его необходимо обосновать и представить на утверждение уполномоченному органу государства-члена до начала проведения. Такой подход применим, например, к испытаниям на остаточные растворители и микробиологическую чистоту в отношении твердых лекарственных форм для приема внутрь. При подаче регистрационного досье лекарственного препарата заявитель может располагать лишь ограниченными данными, поэтому такой подход следует реализовывать на пострегистрационном этапе. Если при периодических (выборочных) испытаниях обнаруживаются какие-либо несоответствия утвержденным критериям приемлемости, об этом необходимо надлежащим образом уведомить уполномоченные органы государств-членов. Если эти несоответствия свидетельствуют о необходимости восстановления рутинных испытаний, следует вернуть испытания при выпуске в отношении каждой серии.
2. Критерии приемлемости при выпуске и в течение срока годности (хранения)
11. Подход, связанный с различием критериев приемлемости для спецификаций при выпуске и в течение срока годности (хранения), применим только к лекарственным препаратам. Он предусматривает установление более строгих критериев приемлемости при выпуске лекарственного препарата по сравнению с критериями приемлемости, применяемыми в течение срока годности (хранения). Для применения такого подхода примерами показателей могут служить количественное определение и родственные примеси.
12. Заявитель вправе предусмотреть более строгие собственные пределы критериев приемлемости на момент выпуска, чтобы обеспечить уверенность в том, что качество лекарственного препарата останется в пределах регламентированного критерия приемлемости на протяжении всего срока годности (хранения).
3. Внутрипроизводственные испытания
13. Внутрипроизводственные испытания – испытания, которые допускается проводить в процессе производства активной фармацевтической субстанции и (или) лекарственного препарата, а не в рамках официального комплекса испытаний, проводимых перед выпуском продукции.
14. В спецификации не включают внутрипроизводственные испытания, которые используются для коррекции параметров технологического процесса в пределах диапазона эксплуатации, установленного для данного технологического процесса (например, твердость и хрупкость ядер таблеток, на которые должно быть нанесено покрытие, и масса отдельных таблеток).
15. Определенные испытания, проводимые в процессе производства, могут быть достаточными для подтверждения соответствия требованиям спецификации, если такие же испытания включены в спецификацию, а критерий приемлемости идентичен требованию, установленному в спецификации, применяемой при выпуске, или является более строгим (например, рН раствора).
16. Вместе с тем этот подход необходимо валидировать для доказательства того, что функциональные характеристики планируемого к получению продукта или результаты его испытаний в процессе производства не изменились по отношению к готовой продукции.
4. Вопросы проектирования (дизайна) и разработки
17. Спецификации следует составлять на основании опыта и данных, накопленных в процессе разработки активной фармацевтической субстанции и (или) лекарственного препарата. На основании этих данных можно вносить предложения об исключении, добавлении или замене определенных испытаний. Например, исключить:
а) испытания на микробиологическую чистоту для активных фармацевтических субстанций и (или) твердых лекарственных форм, которые в процессе разработки подтвердили неспособность поддерживать жизнеспособность и рост микроорганизмов (схемы решений № 6 и 8);
б) определение веществ, экстрагируемых из упаковки, если было доказано с воспроизводимыми результатами, что экстрагируемые вещества в лекарственном препарате не обнаруживаются или их содержание удовлетворяет приемлемым критериям и требованиям безопасности;
в) испытание на определение размера частиц в зависимости от значимости для функциональных характеристик продукта (можно проводить в качестве испытания в процессе производства или при выпуске).
18. Испытание на растворение твердых лекарственных форм для приема внутрь с немедленным высвобождением, содержащих хорошо растворимые в воде активные фармацевтические субстанции, допускается заменить на испытание на распадаемость, если в ходе разработки такие лекарственные препараты проявляли постоянное быстрое высвобождение фармацевтической субстанции (схемы решений № 7(1) и 7(2)).
5. Проблемы ограниченности данных по качеству при подаче регистрационного досье лекарственного препарата
19. На момент подачи регистрационного досье лекарственного препарата данные по его качеству могут быть ограниченными, что может повлиять на процесс установления критериев приемлемости.
В связи с этим по мере накопления опыта при производстве определенной активной фармацевтической субстанции и (или) лекарственного препарата может потребоваться пересмотр критериев приемлемости (например, критериев приемлемости содержания специфической примеси). Критерии приемлемости на момент подачи регистрационного досье лекарственного препарата следует устанавливать на основании требований безопасности и эффективности.
20. При наличии на момент утверждения испытаний и критериев приемлемости лишь ограниченных данных по качеству лекарственного препарата ранее утвержденные испытания и критерии приемлемости необходимо пересмотреть по мере накопления сведений, с учетом возможной модификации испытаний и критериев приемлемости. Могут быть установлены как менее, так и более строгие критерии приемлемости.
6. Выпуск по параметрам
21. Выпуск по параметрам как альтернатива рутинным испытаниям при выпуске может быть использован в ограниченных случаях и исключительно при условии получения одобрения от уполномоченного органа (например, замена испытания на стерильность для лекарственных препаратов, подвергающихся заключительной (терминальной) стерилизации, их выпуском по параметрам). В случае замены испытания на стерильность выпуском по параметрам выпуск каждой серии осуществляется на основании удовлетворительных результатов мониторинга определенных параметров (температуры, давления и продолжительности фаз (фазы) терминальной стерилизации в ходе производства лекарственного препарата). Эти параметры, как правило, можно контролировать и измерять с бóльшей точностью, поэтому они более надежны при обеспечении стерильности, чем испытание на стерильность конечного продукта. В программу выпуска по параметрам могут быть включены соответствующие лабораторные испытания (например, использование химического или физического индикатора). Прежде чем внедрять выпуск по параметрам процесс стерилизации необходимо должным образом валидировать. Следует также подтверждать сохранение валидированного состояния посредством проведения ревалидации через установленные интервалы. При осуществлении выпуска по параметрам в спецификацию необходимо включить показатель качества (например, стерильность), контролируемый косвенно, а также ссылку на связанную с ним аналитическую методику.
7. Альтернативные методики испытаний
22. Альтернативные методики испытаний – методики, которые допускается использовать для определения показателя качества, если они позволяют контролировать качество активной фармацевтической субстанции и (или) лекарственного препарата в той же степени, что и официально утвержденная методика, или в более высокой степени. Например, для таблеток, которые, как было доказано, не разлагаются в процессе производства, в целях выпускающего контроля качества допускается использовать спектрофотометрическую методику, а не официально утвержденную хроматографическую. Тем не менее в целях подтверждения соответствия критериям приемлемости в течение срока годности (хранения) лекарственного препарата необходимо использовать хроматографическую методику.
8. Фармакопейные испытания и критерии приемлемости
23. В Фармакопее Союза, а при отсутствии в ней в фармакопеях государств-членов приведены определенные методики (далее – фармакопейные методики) или ссылки на них. Во всех случаях (если целесообразно) следует использовать фармакопейные методики.
9. Развивающиеся технологии
24. Поскольку непрерывно разрабатываются новые аналитические технологии и вносятся изменения в существующие, следует использовать развивающиеся технологии, если они позволяют обеспечить дополнительную гарантию качества или их применение обосновано иными причинами с точки зрения обеспечения качества или безопасности лекарственного препарата.
10. Влияние активной фармацевтической субстанции на спецификации лекарственного препарата
25. Как правило, нет необходимости проводить испытания лекарственного препарата по показателям качества, характерным исключительно для активных фармацевтических субстанций. Например, лекарственный препарат не требуется испытывать на наличие примесей, контроль которых осуществляется в активной фармацевтической субстанции и которые связаны с процессом синтеза, а не являются продуктами деградации. Более подробные сведения приведены в правилах по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, утверждаемых Евразийской экономической комиссией (далее – Комиссия).
11. Стандартный образец
26. Качество стандартного образца должно соответствовать его назначению. Стандартный образец часто характеризуют и оценивают на пригодность для целевого назначения с помощью дополнительных методик и методов. В стандартных образцах активной фармацевтической субстанции, предназначенных для использования в испытаниях на количественное определение, необходимо надлежащим образом идентифицировать и (или) контролировать примеси и определять чистоту с помощью количественной методики.
V. Требования к спецификациям
1. Спецификации: определение и обоснование
Определение спецификаций
27. В дополнение к испытаниям при выпуске спецификация может содержать перечень испытаний в процессе производства, периодических (выборочных) испытаний и прочих испытаний, которые не всегда проводятся посерийно. В подобных ситуациях заявитель должен указать, какие испытания проводятся посерийно, а какие – нет. При этом необходимо указать и обосновать выбор и частоту испытаний. Активная фармацевтическая субстанция и (или) лекарственный препарат должны удовлетворять критериям приемлемости, если они подвергаются испытаниям. Следует отметить, что внесение изменений в спецификации после утверждения регистрационного досье лекарственного препарата может потребовать предварительной экспертизы уполномоченным органом государства-члена.
Обоснование спецификаций
28. При первичном составлении спецификации необходимо обосновать каждую предлагаемую аналитическую методику и каждый критерий приемлемости. При обосновании необходимо ссылаться на соответствующие данные по разработке, требования Фармакопеи Союза, а при отсутствии в ней соответствующих данных – на требования фармакопей государств-членов, результаты испытаний активных фармацевтических субстанций и (или) лекарственных препаратов, использованных в токсикологических и клинических исследованиях, а также на результаты исследований ускоренных и долгосрочных испытаний стабильности. Кроме того, необходимо учитывать приемлемые диапазоны вероятной вариабельности аналитической методики и вероятной вариабельности процесса производства лекарственного препарата.
29. Могут быть применимыми и приемлемыми иные подходы, не описанные в настоящих Требованиях. Применение таких альтернативных подходов требует обоснования заявителем. При обосновании спецификаций необходимо руководствоваться данными, полученными по результатам синтеза активной фармацевтической субстанции и (или) процесса производства лекарственного препарата. При этом в качестве обоснования можно рассматривать теоретически допустимые пределы для конкретной методики или конкретного критерия приемлемости. Однако независимо от применяемого подхода основополагающими являются экспериментальные фактические результаты.
30. При составлении и обосновании спецификаций следует учитывать результаты испытаний серий, включенных в программу испытаний стабильности, а также серий, полученных при масштабировании (валидации) процесса, необходимо уделять особое внимание исходным сериям, используемым для испытаний стабильности. Если планируется использование нескольких производственных площадок, то при первоначальном выборе основных испытаний и критериев приемлемости целесообразно учитывать данные, полученные на этих площадках. Это необходимо в случае, если имеется недостаточный первоначальный опыт производства активной фармацевтической субстанции и (или) лекарственного препарата на данной производственной площадке. Если для выбора испытаний и критериев приемлемости используется 1 репрезентативная производственная площадка, то продукт, произведенный на остальных площадках, должен соответствовать этим критериям.
31. При обосновании отдельных критериев приемлемости рекомендуется представление результатов испытаний в графической форме (в частности, значений количественного содержания активной фармацевтической субстанции и родственных примесей). При таком представлении результатов необходимо включить в спецификацию данные, полученные на стадии разработки, а также имеющиеся данные о стабильности серий активной фармацевтической субстанции или лекарственного препарата, произведенного с помощью предлагаемого процесса производства лекарственного препарата.
32. При обосновании исключения испытания из спецификации необходимо руководствоваться данными о разработке и валидации процесса производства лекарственного препарата (если применимо).
2. Универсальные испытания (критерии)
33. При выполнении требований, указанных в пунктах 34 – 36 настоящих Требований, необходимо также учитывать требования Руководства по валидации аналитических методик проведения испытаний лекарственных средств, утвержденного Решением Коллегии Евразийской экономической комиссии от 17 июля 2018 г. № 113.
Фармацевтические субстанции
34. Следующие испытания и критерии приемлемости применимы ко всем активным фармацевтическим субстанциям:
а) описание – качественная характеристика физического состояния (например, твердое вещество, жидкость) и цвета активной фармацевтической субстанции. Если одно из этих свойств изменяется при хранении, такое изменение необходимо изучить и принять необходимые меры;
б) идентификация – испытания идентификации должны давать возможность наилучшим образом различать соединения с близкородственной структурой, которые могут присутствовать в активной фармацевтической субстанции и (или) лекарственном препарате с высокой долей вероятности. Испытания идентификации должны быть специфичными в отношении активной фармацевтической субстанции (например, инфракрасная спектроскопия). Идентификация только с помощью хроматографического времени удержания не считается специфичной. Вместе с тем использование
2 хроматографических методик, в которых разделение детектируемых веществ основано на различных принципах, или объединение испытаний в 1 методику (например, ВЭЖХ/УФ на диодной матрице, ВЭЖХ/МС или ГХ/МС) является приемлемым. Если активная фармацевтическая субстанция представляет собой соль, испытание идентификации должно быть специфичным в отношении каждого из ионов. Может быть достаточным испытание, специфичное в отношении самой соли. Для оптически активных фармацевтических субстанций может быть необходимо проведение специфичного испытания для их идентификации или проведение количественного определения, специфичного в отношении хирального соединения;
в) количественное определение – для определения содержания в анализируемом образце активной фармацевтической субстанции необходимо включить в спецификацию специфичную методику, позволяющую получать стабильные результаты. Во многих случаях допускается использовать одну и ту же методику (например, ВЭЖХ) как для количественного определения активной фармацевтической субстанции, так и для определения содержания примесей. Если представлено обоснование использования неспецифичной методики количественного определения, для достижения общей специфичности необходимо использовать другие подтверждающие методики испытаний. Например, если для количественного определения активной фармацевтической субстанции используется титрование, необходимо использовать комбинацию количественного определения и подходящего испытания на содержание примеси;
г) примеси – в спецификации указывают органические и неорганические примеси, остаточные растворители. Экстраполяция значимых пределов содержания примесей на основе данных, полученных в ходе разработки, описана в схеме решений № 1. Поскольку на момент подачи регистрационного досье лекарственного препарата данные для оценки постоянства процесса могут быть недостаточными, нецелесообразно устанавливать критерии приемлемости, которые охватывают только данные анализа серий, имеющиеся на момент подачи регистрационного досье лекарственного препарата.
Лекарственные препараты
35. Следующие испытания и критерии приемлемости применимы ко всем лекарственным препаратам:
а) описание – в спецификации приводится описание качественных характеристик лекарственной формы (например, размер, форма и цвет). Если одно из этих свойств изменяется в ходе процесса производства лекарственного препарата или при хранении лекарственного препарата, такое изменение необходимо изучить и принять необходимые меры. Критерии приемлемости должны включать в себя внешний вид лекарственного препарата. Если при хранении лекарственного препарата его внешний вид изменяется по цвету, то в спецификацию требуется включение методики количественной оценки;
б) идентификация – испытания должны устанавливать подлинность активной фармацевтической субстанции в лекарственном препарате, а также позволять различать соединения с близкородственной структурой, которые могут присутствовать с высокой долей вероятности в составе активной фармацевтической субстанции и (или) лекарственном препарате. Испытания идентификации должны быть специфичными в отношении активной фармацевтической субстанции (например, инфракрасная спектроскопия). Идентификация только с помощью хроматографического времени удержания не считается специфичной. Как правило, использование 2 хроматографических методик, в которых разделение основано на различных принципах, или объединение испытаний в 1 методику (например, ВЭЖХ/УФ на диодной матрице, ВЭЖХ/МС или ГХ/МС) является приемлемым;
в) количественное определение – в отношении всех лекарственных препаратов для определения содержания в спецификацию необходимо включить специфичную методику количественного определения, позволяющую получать стабильные результаты. Во многих случаях допускается использовать одну и ту же методику (например, ВЭЖХ) как для количественного определения активной фармацевтической субстанции, так и для определения содержания примесей. Для количественного определения активной фармацевтической субстанции в лекарственных препаратах могут быть использованы результаты испытаний на однородность содержания, если методы, используемые для определения однородности содержания, являются также приемлемыми для количественного определения. Если представлено обоснование использования неспецифичной методики количественного определения, для достижения общей специфичности необходимо использовать другие подтверждающие аналитические методики. Например, если для количественного определения активной фармацевтической субстанции используется титрование, необходимо использовать комбинацию количественного определения и подходящего испытания на содержание примеси. Если в случае использования неспецифичной методики количественного определения имеются данные о влиянии вспомогательных веществ на результаты анализа, необходимо использовать специфичную методику;
г) примеси – в спецификации указываются органические и неорганические примеси, продукты деградации, остаточные растворители. Органические примеси, образующиеся при деградации активной фармацевтической субстанции, и примеси, образующиеся в процессе производства лекарственного препарата, подлежат контролю. Следует установить приемлемые пределы содержания индивидуальных специфицированных продуктов деградации, которые могут представлять собой как идентифицированные, так и неидентифицированные продукты деградации, а также пределы суммарного содержания продуктов деградации. Примеси, образующиеся в процессе синтеза активной фармацевтической субстанции, как правило, контролируются на стадии ее испытания и не включаются в предел содержания суммы примесей. Если технологическая примесь является также продуктом деградации, ее содержание необходимо контролировать и включать в предел содержания суммы продуктов деградации. Если с помощью надлежащей аналитической методики однозначно доказано, что активная фармацевтическая субстанция, входящая в состав конкретного лекарственного препарата, в конкретных условиях хранения, указанных в регистрационном досье лекарственного препарата, не подвергается деградации, то после регистрации такого лекарственного препарата по согласованию с уполномоченным органом допускается сократить объем испытания на продукты деградации или исключить его из спецификации.
36. В схеме решений № 2 описана экстраполяция значимых пределов содержания продуктов деградации на основе данных, полученных в процессе разработки. На момент подачи регистрационного досье лекарственного препарата данных для оценки постоянства процесса производства лекарственного препарата, как правило, недостаточно. В связи с этим нецелесообразно устанавливать критерии приемлемости, которые охватывают только данные анализа серий, имеющиеся на момент подачи регистрационного досье лекарственного препарата.
3. Специфичные испытания (критерии)
37. Помимо универсальных испытаний, указанных в пунктах 34 – 36 настоящих Требований, в отношении отдельных активных фармацевтических субстанций и (или) лекарственных препаратов требуется проведение следующих дополнительных испытаний. Если испытание оказывает влияние на контроль качества серий активной фармацевтической субстанции и (или) лекарственного препарата, в спецификацию необходимо включить индивидуальные испытания (критерии). В отдельных случаях (например, после накопления данных об изменениях качества лекарственного препарата) может потребоваться проведение иных испытаний.
Специфические испытания (критерии) для фармацевтических субстанций
38. Следующие специфические испытания (критерии) применимы ко всем активным фармацевтическим субстанциям:
а) физико-химические свойства. Включают в себя в том числе pH водного раствора, температуру (диапазон температур) плавления и показатель преломления. Методики, используемые для определения указанных свойств, как правило, уникальны и не требуют сложных измерений (например, капиллярный метод определения температуры плавления, рефрактометрия по Аббе). Испытания, выполняемые с целью подтверждения физико-химических свойств, необходимо определять на основании физических и химических свойств активной фармацевтической субстанции и ее целевого назначения;
б) размер частиц. Для некоторых активных фармацевтических субстанций, предназначенных для использования в твердых лекарственных препаратах или суспензиях, размер частиц может оказывать значительное влияние на скорость растворения, биодоступность и (или) стабильность. В таких случаях следует установить критерии приемлемости и проводить испытание по определению размера частиц с использованием соответствующей методики. В схеме решений № 3 представлены дополнительные пояснения в отношении имеющихся случаев проведения испытания по определению размера частиц;
в) полиморфные формы. Некоторые активные фармацевтические субстанции существуют в разных кристаллических формах, которые различаются по своим физическим свойствам. Полиморфизм может также включать в себя продукты сольватации и гидратации (псевдополиморфы) и аморфные формы. Различия в профиле указанных форм могут в некоторых случаях оказывать влияние на качество и функциональные характеристики лекарственных препаратов. Если существуют различия с доказанным влиянием на функциональные характеристики лекарственного препарата, его биодоступность или стабильность, то в спецификации необходимо указать соответствующее состояние твердого вещества. Физико-химические измерения и методы обычно используются для определения наличия нескольких полиморфных форм (например, точка плавления (включая микроскопию при высокой температуре), инфракрасная спектроскопия (ИК) для веществ в твердом состоянии, порошковая рентгеновская дифракция, методики термического анализа (включая дифференциально-сканирующую калориметрию (ДСК), термогравиметрический анализ (ТГА) и дифференциально-термический анализ (ДТА)), Рамановская спектроскопия, световая микроскопия и спектроскопия ядерного магнитного резонанса (ЯМР) для веществ в твердом состоянии). В схемах решений № 4 (1) – 4 (3) представлены дополнительные пояснения, когда и как следует контролировать и проверять полиморфные формы. Указанные схемы решений следует применять последовательно. В схемах решений № 4 (1) и 4 (2) рассматривается, проявляет ли активная фармацевтическая субстанция полиморфизм и могут ли различные полиморфные формы повлиять на функциональные характеристики лекарственного препарата. Схему решений № 4 (3) следует использовать в том случае, если активная фармацевтическая субстанция проявляет полиморфизм и он оказывает влияние на функциональные характеристики лекарственного препарата. С помощью схемы решений № 4 (3) можно проанализировать возможность изменения профиля полиморфных форм в лекарственном препарате, а также способность такого изменения оказывать влияние на его функциональные характеристики.
Как правило, очень трудно технически измерить полиморфные изменения в лекарственных препаратах. В целях контроля функциональных характеристик лекарственного препарата, как правило, используются косвенные испытания (например, растворение) (схема решений № 4 (3)), а определение содержания полиморфных форм и установление критериев приемлемости для такого испытания следует проводить только в крайнем случае;
г) испытания для хиральных активных фармацевтических субстанций. Если активная фармацевтическая субстанция представляет собой преимущественно 1 энантиомер, а содержание противоположного энантиомера ниже квалификационных и идентификационных пороговых значений, приведенных в правилах по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, утверждаемых Комиссией, то противоположный энантиомер не определяется по причине практической сложности определения при его низком уровне содержания. Однако такую примесь в хиральной активной фармацевтической субстанции и соответствующих ей лекарственных препаратах следует контролировать в соответствии с правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях. В схеме решений № 5 представлено обобщение условий, требующих проведения испытаний идентификации хиральных соединений, испытаний на определение содержания примесей и на количественное определение как для активных фармацевтических субстанций, так и для лекарственных препаратов в соответствии со следующими концепциями.
Активные фармацевтические субстанции: примеси. Контроль противоположного энантиомера хиральных активных фармацевтических субстанций, разработанных в качестве одиночного энантиомера, проводится таким же образом, как и в отношении других примесей. Однако технические ограничения могут препятствовать использованию тех же пределов квантификации количественного содержания или квалификационных пределов. Используя соответствующее обоснование, следует также показать надежность контроля с помощью подходящих испытаний исходного сырья или промежуточной продукции.
Количественное определение. В спецификацию должно быть включено испытание на количественное определение активной фармацевтической субстанции, позволяющее селективно устанавливать содержание энантиомеров в активной фармацевтической субстанции. Для выполнения этих условий считается приемлемым использование специфичной для хирального соединения методики количественного определения с соответствующими методами контроля энантиомерной примеси.
Идентификация. Для активной фармацевтической субстанции, разработанной в качестве одиночного энантиомера, испытания идентификации должны обеспечивать возможность различать каждый энантиомер и рацемическую смесь энантиомеров. Выделяются 2 случая, при которых активная фармацевтическая субстанция, представляющая собой рацемическую смесь, требует проведения испытания идентификации стереоизомеров при проведении испытаний при выпуске (испытания на пригодность) в случае высокой вероятности замещения рацемата энантиомером или оснований для избирательной кристаллизации, которая может привести к непреднамеренному образованию нерацемической смеси.
Лекарственный препарат: продукты деградации. При отсутствии подтверждения того, что в процессе производства лекарственной формы и ее хранении рацемизация является незначительной, необходимо осуществлять контроль содержания в лекарственном препарате второго энантиомера.
Количественное определение. Если доказано, что в процессе производства лекарственной формы и ее хранении рацемизация является незначительной, допускается использовать методику количественного определения, неспецифичную в отношении хирального соединения. В иных случаях необходимо использовать методику, специфичную в отношении хирального соединения, или
в качестве альтернативы комбинацию неспецифичной методики и валидированной методики контроля содержания противоположного энантиомера.
Идентификация. В спецификацию лекарственного препарата на момент выпуска, как правило, не включается стереоспецифическое испытание идентификации. Если при производстве лекарственной формы и ее хранении рацемизация является незначительной, то стереоспецифичное испытание идентификации целесообразно включить в спецификацию активной фармацевтической субстанции. Если в лекарственной форме происходит рацемизация, то для подтверждения подлинности используется хиральная методика количественного определения или испытание на энантиомерную примесь в лекарственном препарате;
д) содержание воды. Данное испытание проводится в случае, если активная фармацевтическая субстанция гигроскопична, или влага вызывает ее деградацию, или активная фармацевтическая субстанция представляет собой стехиометрический гидрат. Критерии приемлемости могут быть обоснованы с помощью данных об эффектах гидратации или абсорбции влаги. В некоторых случаях достаточно использовать методику определения потери в массе при высушивании, но предпочтительно использовать методику обнаружения, специфичную в отношении воды (например, титрование по методу Фишера);
е) неорганические примеси. На основе данных, полученных при разработке, и основываясь на знании процесса производства лекарственного препарата, следует определить необходимость включения испытаний и критериев приемлемости на неорганические примеси (например, катализаторы). Методики и критерии приемлемости для сульфатной золы (общей золы) должны соответствовать требованиям Фармакопеи Союза, а при отсутствии в ней соответствующих данных – требованиям фармакопей государств-членов. Прочие неорганические примеси допускается определять с помощью других подходящих методик (например, атомно-абсорбционной спектрометрии);
ж) микробиологическая чистота. В спецификации может потребоваться указание общего содержания аэробных микроорганизмов, общего количества дрожжевых и плесневых грибов, а также информации о полном отсутствии определенных видов бактерий, наличие которых в лекарственном препарате не допускается (например, Staphylococcus aureus, Escherichia coli, Salmonella spp., Pseudomonas aeruginosa). Присутствие микроорганизмов необходимо определить с использованием фармакопейных методик. Разновидность микробиологических испытаний и критериев приемлемости следует определять на основании свойств активной фармацевтической субстанции, способа производства и предлагаемого применения лекарственного препарата. Например, в отношении стерильных активных фармацевтических субстанций целесообразно проведение испытания на стерильность, а испытание на эндотоксины целесообразно проводить с активной фармацевтической субстанцией, используемой для приготовления инъекционных форм лекарственных препаратов. В схеме решений № 6 представлены дополнительные пояснения, в каких случаях следует включать в спецификацию испытания на микробиологическую частоту.
Специфические испытания (критерии) для лекарственных препаратов
39. Указанный в пунктах 40 и 41 настоящих Требований перечень представляет собой репрезентативный список лекарственных препаратов, а также разновидностей испытаний и критериев приемлемости, которые целесообразно включать в их спецификацию. В перечне рассматриваются твердые и жидкие лекарственные формы для приема внутрь и парентеральные лекарственные формы (малого и большого объемов). Указанные в пунктах 40 и 41 настоящих Требований принципы могут быть применимы к другим лекарственным формам.
Таблетки, таблетки, покрытые оболочкой, и капсулы
40. К мягким капсулам и гранулам могут быть применимы следующие из описанных испытаний:
а) растворение. В спецификацию твердых лекарственных форм для приема внутрь, как правило, включаются испытания для определения высвобождения активной фармацевтической субстанции из лекарственного препарата. В отношении лекарственных форм с немедленным высвобождением, как правило, достаточно «одноточечных» измерений. В отношении лекарственных форм с модифицированным высвобождением необходимо подобрать подходящие условия испытаний и методики отбора проб. Например, в отношении лекарственных форм с пролонгированным высвобождением отбор проб необходимо осуществлять в нескольких временны́х точках, а в отношении лекарственных форм с отсроченным высвобождением целесообразно проводить двухэтапное испытание (последовательно или параллельно, с использованием различных сред в зависимости от ситуации). В таких случаях при выборе испытаний и установлении критериев приемлемости необходимо принимать во внимание популяцию лиц, которым предназначается лекарственный препарат (например, пожилые лица, страдающие ахлоргидрией).
В некоторых случаях испытание на растворение допускается заменить испытанием на распадаемость (схема решений № 7 (1)).
Если изменение скорости растворения лекарственных препаратов с немедленным высвобождением может значительно повлиять на биодоступность, рекомендуется предусмотреть такие условия испытаний, которые смогут выявлять серии с неприемлемой биодоступностью. Если изменения в составе или в параметрах процесса производства лекарственного препарата существенно влияют на растворение и такие изменения не контролируются с помощью другого установленного в спецификации метода, то целесообразно предусмотреть условия испытания на растворение, которые позволят распознать подобные изменения (схема решений № 7 (2)).
Если растворение существенно влияет на биодоступность, то необходимо установить критерии приемлемости для испытания на растворение, позволяющие отбраковать серии с неприемлемой биодоступностью. В иных случаях необходимо выбрать такие условия испытания и критерии приемлемости, которые позволят выбрать серии лекарственного препарата, пригодные для применения лекарственного препарата в медицинских целях (схема решений № 7 (2)).
В отношении лекарственных препаратов с пролонгированным высвобождением различных составов, проявляющих различную скорость высвобождения, в целях установления критериев приемлемости допускается для их обоснования использовать in vivo (in vitro) корреляцию при наличии данных о биодоступности у человека таких лекарственных препаратов. Если данные о биодоступности у человека отсутствуют и невозможно подтвердить, что высвобождение активной фармацевтической субстанции не зависит от условий испытаний in vitro, то критерии приемлемости устанавливают на основании имеющихся результатов испытаний серий. Допустимые отклонения средней скорости высвобождения во всех временных точках не должны превышать ± 10 % от заявленного содержания активной фармацевтической субстанции (т. е. общая вариабельность не более 20 %: требование 50 ± 10 % означает, что приемлемый диапазон равен 40 – 60 %), если только более широкий диапазон не обоснован исследованием биоэквивалентности (схема решений № 7 (3));
б) распадаемость. В отношении быстрорастворяющихся лекарственных препаратов (растворение > 80 % за 15 минут при pH, равном 1,2, 4,0 и 6,8), содержащих хорошо растворимые активные фармацевтические субстанции в физиологическом диапазоне значений pH (доза растворяется в среде объемом менее 250 мл при pH от 1,2 до 6,8), испытание на растворение допускается заменить испытанием на распадаемость. Проведение испытания на распадаемость наиболее целесообразно, если была установлена взаимосвязь с растворением или было показано, что определение распадаемости подходит лучше, чем испытание на растворение. В таких случаях испытание на растворение может не потребоваться. При выборе испытаний на растворение или распадаемость предполагается, что для подтверждения надежности состава лекарственного препарата и производственного процесса будет представлена информация о разработке (схема решений № 7 (1));
в) испытания на твердость и (или) истираемость. Если испытания на твердость и (или) истираемость выполняются в процессе производства лекарственного препарата, то эти показатели качества не требуется включать в спецификацию. Если твердость и истираемость оказывают критическое влияние на качество лекарственного препарата (например, жевательных таблеток), то в спецификацию необходимо включить соответствующие испытания и критерии приемлемости;
г) однородность единиц дозирования (однородность дозирования, определяемая методом вариации масс, и однородность дозирования, определяемая методом однородности содержания активной фармацевтической субстанции в единице дозированной формы). При испытаниях необходимо использовать фармакопейные методики.
В спецификацию включают одно из этих испытаний. Если применимо, указанные испытания проводятся в процессе производства лекарственного препарата; при этом в спецификацию необходимо включить критерии приемлемости. Если отклонения в массе лекарственных препаратов превышают пороговое значение, при котором можно определять однородность содержания путем определения отклонения в массе, заявители в ходе разработки должны удостовериться, что гомогенность лекарственного препарата является удовлетворительной;
д) содержание воды. При необходимости в спецификацию включается испытание на содержание воды. Критерии приемлемости допускается обосновывать данными о влиянии гидратации или абсорбции воды лекарственным препаратом. В некоторых случаях достаточно использовать методику определения потери в массе при высушивании, но предпочтительнее использовать методику обнаружения, специфичную в отношении воды (например, титрование по методу Фишера);
е) микробиологическая чистота. Испытания на микробиологическую чистоту необходимы для подтверждения выполнения Правил надлежащей производственной практики и для обеспечения качества лекарственного препарата. Такие испытания в отношении лекарственных препаратов допускается не проводить, если выполняются 2 условия:
компоненты лекарственного препарата подвергаются испытаниям до начала производства;
в процессе производства лекарственного препарата по результатам валидационных исследований отсутствует значительный риск микробной контаминации или пролиферации.
Положения настоящих Требований распространяются на вспомогательные вещества и на лекарственные препараты. В обоих случаях допускается использовать подход, основанный на проведении выборочных испытаний (если он применим) (схема решений № 6).
В спецификацию необходимо включить критерии приемлемости для содержания общего количества аэробных микроорганизмов, общего количества дрожжевых и плесневых грибов, а также информацию о полном отсутствии определенных видов бактерий, наличие которых в лекарственном препарате не допускается (например, Staphylococcus aureus, Escherichia coli, Salmonella spp., Pseudomonas aeruginosa). Микробиологическую чистоту следует определять с помощью подходящих фармакопейных методик, при этом частота отбора проб или временные точки производственного процесса должны быть обоснованы данными и накопленным опытом. При выборе вида испытаний на микробиологическую чистоту и критериев приемлемости следует учитывать природу активной фармацевтической субстанции, способ производства и назначение лекарственного препарата. При должном научном обосновании в отношении жидких лекарственных форм для приема внутрь допускается не проводить испытания на микробиологическую чистоту. Дополнительные пояснения о том, в каких случаях следует проводить микробиологические испытания, представлены в схеме решений № 8.
Жидкие лекарственные формы для приема внутрь
41. Для жидких лекарственных форм для приема внутрь и порошков, предназначенных для приготовления жидких лекарственных форм для приема внутрь, применяются следующие испытания:
а) однородность единиц дозирования. Понятие включает в себя как однородность дозирования, определяемую методом вариации масс, так и однородность дозирования, определяемую методом однородности содержания активной фармацевтической субстанции в лекарственном препарате. При испытаниях необходимо использовать фармакопейные методики. В спецификацию, как правило, включают одно из этих испытаний, но не оба одновременно. Если применимо, указанные испытания проводятся в процессе производства лекарственного препарата, при этом в спецификацию необходимо включить критерии приемлемости. Это правило распространяется на лекарственные средства как в однодозовых, так и в многодозовых упаковках. Если отклонения в массе лекарственных препаратов превышают пороговое значение, при котором можно определять однородность содержания путем определения отклонения в массе, заявители в ходе разработки должны удостовериться, что гомогенность лекарственного препарата удовлетворительна.
Единица дозирования – это доза, применяемая пациентом за 1 прием. Если фактически применяемая пациентом доза контролируется, ее можно определить напрямую или с помощью расчета: путем деления общей измеренной массы или объема лекарственного препарата на предполагаемое число доз. Если устройство для дозирования (например, медицинская пипетка или пробка-капельница для флаконов) является составной частью упаковки, то в целях определения дозы необходимо использовать это устройство. В противном случае необходимо использовать стандартные единицы измерения объема. Выбор дозирующего устройства определяют в ходе разработки. Для порошков, предназначенных для растворения, как правило, считается приемлемым испытание на однородность массы;
б) pH. Если применимо, необходимо представить критерии приемлемости относительно диапазона pH и обоснования его выбора;
в) микробиологическая чистота. Испытания на микробиологическую чистоту необходимы для подтверждения выполнения Правил надлежащей производственной практики и для обеспечения качества лекарственного препарата. Такие испытания в отношении лекарственных препаратов допускается не проводить если выполняются 2 условия:
компоненты лекарственного препарата подвергаются испытаниям до начала производства;
в процессе производства лекарственного препарата по результатам валидационных исследований отсутствует значительный риск микробной контаминации или пролиферации.
Положения настоящих Требований распространяются как на вспомогательные вещества, так и на лекарственные препараты. В обоих случаях допускается использовать подход, основанный на проведении выборочных испытаний (если он применим) (схема решений № 6).
В спецификации необходимо указать критерии приемлемости для содержания общего количества аэробных микроорганизмов, общего количества дрожжевых и плесневых грибов, а также информацию о полном отсутствии определенных видов бактерий, наличие которых в лекарственном препарате не допускается (например, Staphylococcus aureus, Escherichia coli, Salmonella spp., Pseudomonas aeruginosa). Микробиологическую чистоту следует определять с помощью подходящих фармакопейных методик, при этом частота отбора проб или временные точки производственного процесса должны быть обоснованы данными и накопленным опытом. При выборе вида испытаний на микробиологическую чистоту и критериев приемлемости следует учитывать природу активной фармацевтической субстанции, способ производства и назначение лекарственного препарата. При должном научном обосновании в отношении жидких лекарственных форм для приема внутрь допускается не проводить испытания на микробиологическую чистоту. Дополнительные пояснения о том, в каких случаях следует проводить микробиологические испытания, представлены в схеме решений № 8;
г) содержание антимикробного консерванта. В отношении жидких лекарственных форм для приема внутрь, требующих добавления антимикробного консерванта, необходимо установить критерии приемлемости относительно его содержания. При выборе критериев приемлемости содержания антимикробного консерванта следует руководствоваться количеством антимикробного консерванта, необходимым для поддержания микробиологической чистоты лекарственного препарата на всех этапах его применения и в течение всего срока годности (хранения). Необходимо, используя фармакопейные методики, доказать эффективность антимикробного консерванта в отношении задержки роста микроорганизмов при самой низкой указанной в спецификации концентрации. Испытание на содержание антимикробного консерванта, как правило, необходимо проводить при выпуске серии. В некоторых случаях взамен испытания на момент выпуска может быть достаточно проведения испытания в процессе производства лекарственного препарата. В случае если испытание на содержание антимикробного консерванта является внутрипроизводственным, его критерии приемлемости также необходимо включить в спецификацию. Несмотря на то, что химическое испытание на содержание антимикробного консерванта является стандартным показателем, включаемым в спецификацию, в ходе разработки, масштабирования и на протяжении срока годности (хранения) (например, при испытании на стабильность, описанном в Решении Коллегии Евразийской экономический комиссии от 10 мая 2018 г. № 69 «Об утверждении Требований к исследованию стабильности лекарственных препаратов и фармацевтических субстанций») должна быть доказана эффективность антимикробного консерванта;
д) одержание антиоксидантов. Испытание на содержание антиоксидантов необходимо проводить при выпуске серии. При определенных обстоятельствах на основании данных по разработке лекарственного препарата и результатов испытания стабильности допускается не проводить испытание на конец срока годности (хранения), а взамен испытания при выпуске провести испытание в процессе производства лекарственного препарата (при наличии соответствующего обоснования). Если испытание на содержание антиоксидантов проводится в процессе производства лекарственного препарата, то критерии приемлемости необходимо включить в спецификацию. Если испытание на содержание антиоксидантов проводят исключительно при выпуске, то при внесении изменений в технологический процесс или систему упаковки (укупорки) для применения такого подхода следует провести повторное испытание;
е) экстрагируемые вещества. Если данные, полученные в ходе разработки и испытаний стабильности активной фармацевтической субстанции и (или) лекарственного препарата, свидетельствуют о том, что содержание экстрагируемых из системы упаковки (укупорки) веществ постоянно ниже уровней содержания, которые являются приемлемыми и безопасными, то допускается исключить испытание на их содержание из спецификации. При внесении изменений в систему упаковки (укупорки) или в состав лекарственного препарата этот подход необходимо пересмотреть. Если данные свидетельствуют о необходимости проведения испытаний и включения критериев приемлемости для веществ, экстрагируемых из компонентов системы упаковки (укупорки) (например, из резиновых пробок, прокладок в колпачке, пластиковых флаконов и т. д.), то в отношении лекарственных препаратов, первичная упаковка которых изготовлена не из стекла или помещенных в стеклянные флаконы с укупорочными элементами, изготовленными не из стекла, целесообразно проведение испытаний на экстрагируемые вещества и установление критериев приемлемости для этих испытаний. Необходимо перечислить компоненты системы упаковки (укупорки) и представить данные по этим компонентам, начиная с наиболее ранней стадии разработки;
ж) содержание спирта. Если в соответствии с Требованиями к маркировке лекарственных средств для медицинского применения и ветеринарных лекарственных средств, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 76, в информации на этикетке указывается содержание спирта, то его необходимо включить в спецификацию. Содержание спирта определяют методом прямого количественного определения или расчетным методом;
з) растворение. В отношении суспензий для приема внутрь и сухих порошкообразных лекарственных препаратов, подлежащих ресуспендированию, в дополнение к описанным выше показателям качества в спецификацию целесообразно включать (например, в отношении нерастворимых активных фармацевтических субстанций) испытание на растворение и критерии приемлемости. Испытание на растворение следует проводить при выпуске серии. При наличии обоснований, полученных в ходе разработки лекарственного препарата, испытание может проводиться в процессе производства лекарственного препарата. Приборы для проведения испытания, срéды и условия испытания должны быть фармакопейными, в противном случае необходимо представить соответствующее обоснование. Методики растворения с использованием фармакопейных или нефармакопейных условий и приборов необходимо валидировать.
В отношении лекарственных форм с немедленным высвобождением, как правило, достаточно «одноточечных» определений. В отношении лекарственных форм с модифицированным высвобождением отбор проб необходимо осуществлять во множестве точек через соответствующие временные интервалы. Критерии приемлемости необходимо устанавливать на основании наблюдаемого интервала отклонений; при этом следует учитывать профили растворения серий, оказавшихся по результатам исследования in vivo приемлемыми. При выборе между испытанием на растворение и испытанием на распределение частиц по размеру необходимо принимать во внимание данные, полученные в ходе разработки;
и) распределение частиц по размеру. В спецификацию целесообразно включить количественные критерии приемлемости и методики определения размера частиц. При выборе между испытанием на растворение и испытанием на распределение частиц по размеру необходимо принимать во внимание данные, полученные в ходе разработки. Испытание на распределение частиц по размеру следует осуществлять при выпуске. При наличии обоснований в виде данных, полученных в ходе разработки лекарственного препарата, испытание может быть проведено в процессе производства лекарственного препарата. Если в ходе разработки доказано, что лекарственный препарат постоянно характеризуется быстрым высвобождением активной фармацевтической субстанции, то допускается рассмотреть возможность исключения испытания на распределение размера частиц по размеру из спецификации.
При наличии обоснований испытание на растворение допускается заменить испытанием распределения частиц по размеру. Критерии приемлемости должны включать распределение частиц по размеру, которое выражают как процент частиц, имеющих размер в данном диапазоне, от общего числа частиц. Необходимо четко установить предельные значения для среднего, верхнего и (или) нижнего размера частиц.
Критерии приемлемости необходимо устанавливать на основании наблюдаемого диапазона отклонений. При этом следует учитывать профили растворения серий, оказавшихся по результатам исследования in vivo приемлемыми, а также предполагаемое применение лекарственного препарата. В ходе разработки необходимо изучить возможность увеличения размеров частиц. Результаты указанных исследований следует учитывать при выборе критериев приемлемости;
к) ресуспендируемость. В отношении суспензий, характеризующихся оседанием частиц дисперсной фазы при хранении (седиментацией), целесообразно установить критерии приемлемости в отношении ресуспендируемости. Подходящей методикой может служить взбалтывание. В спецификации указывается методика проведения испытания (механическим или ручным способом). Следует четко установить время, необходимое для ресуспендирования при использовании указанной методики. Для обоснования возможности проведения выборочных испытаний серий или исключения данного показателя из спецификации может быть достаточно данных, полученных в ходе разработки лекарственного препарата;
л) реологические свойства. Для относительно вязких растворов и суспензий в спецификацию целесообразно включить испытания реологических свойств (вязкость, удельную плотность). Необходимо указать методику испытания и критерии приемлемости. Для обоснования возможности проведения выборочных испытаний серий или исключения данного показателя из спецификации достаточно данных, полученных в ходе разработки лекарственного препарата;
м) время восстановления. Для сухих порошкообразных лекарственных препаратов, предназначенных для разведения перед применением, необходимо указать критерии приемлемости относительно времени восстановления. Необходимо обосновать выбор растворителя. Для обоснования возможности проведения выборочных испытаний серий или исключения данного показателя из спецификации достаточно данных, полученных в ходе разработки лекарственного препарата;
н) содержание воды. В отношении лекарственных препаратов для приема внутрь, предназначенных для разведения, если применимо, необходимо указать методику и критерий приемлемости содержания воды. Если в ходе разработки лекарственного препарата влияние абсорбированной влаги и гидратной воды хорошо охарактеризовано, то достаточным считается проведение испытания на потерю в массе при высушивании. Следует использовать методику обнаружения, специфичную для воды (например, титрование по методу Фишера).
Лекарственные препараты для парентерального применения
42. Для парентеральных лекарственных препаратов применяются следующие испытания:
а) однородность единиц дозирования. Условия включения испытания в спецификацию приведены в абзаце первом подпункта «а» пункта 41 настоящих Требований. В отношении порошков для восстановления, не содержащих других добавленных активных и вспомогательных веществ, а также для многокомпонентных порошков для восстановления, полученных из истинных растворов, лиофилизированных в конечном контейнере, считается приемлемым испытание на однородность массы;
б) pH. Если применимо, необходимо представить критерии приемлемости по pH и обосновать предлагаемый диапазон;
в) стерильность. Для всех парентеральных лекарственных препаратов необходимо включить методику испытания и критерий приемлемости для оценки стерильности. Если данные, полученные в ходе разработки и валидации, обосновывают выпуск по параметрам, такой подход допускается предложить в отношении лекарственных препаратов, подвергающихся терминальной (заключительной) стерилизации;
г) бактериальные эндотоксины (пирогены). В спецификацию необходимо включить методику испытания и критерий приемлемости в отношении бактериальных эндотоксинов, применяя методику с использованием лизата амебоцитов мечехвоста (ЛАЛ-тест).
При наличии обоснований испытание на эндотоксины допускается заменить испытанием на пирогенность;
д) механические включения. Для парентеральных лекарственных препаратов необходимо предусмотреть надлежащие критерии приемлемости в отношении механических включений. К ним относятся критерии приемлемости для видимых частиц, прозрачности раствора и при необходимости – для невидимых частиц;
е) содержание воды. Для неводных парентеральных лекарственных препаратов и парентеральных лекарственных препаратов, требующих восстановления, следует указать аналитическую методику и критерий приемлемости относительно содержания воды, как это указано в подпункте «н» пункта 41 настоящих Требований;
ж) содержание антимикробного консерванта. Условия включения испытания в спецификацию приведены в подпункте «г» пункта 41 настоящих Требований;
з) содержание антиоксидантов (антиоксидантных консервантов). Условия включения испытания в спецификацию приведены в подпункте «д» пункта 41 настоящих Требований;
и) экстрагируемые вещества. Условия включения испытания в спецификацию приведены в подпункте «е» пункта 41 настоящих Требований;
к) испытание функциональных характеристик систем доставки. Парентеральные лекарственные формы, упакованные в предварительно заполненные шприцы, картриджи-аутоинжекторы или их эквиваленты, необходимо подвергать испытаниям с соответствующими критериями приемлемости в отношении функциональных характеристик системы доставки. К ним относятся контроль проходимости иглы, давление и герметичность укупорки (утечка) и (или) такие параметры, как усилие для снятия винтового колпачка, усилие для перемещения поршня и усилие для приведения в действие инжектора. При определенных обстоятельствах указанные испытания могут быть проведены в процессе производства лекарственного препарата. Для обоснования возможности проведения выборочных испытаний серий или исключения некоторых или всех характеристик из спецификации достаточно данных, полученных в ходе разработки лекарственного препарата;
л) осмолярность. Если на этикетке лекарственного препарата указывается его тоничность, необходимо осуществлять надлежащий контроль осмолярности. Для обоснования проведения этого испытания в процессе производства лекарственного препарата, выборочных испытаний серий или определения этого показателя расчетным методом может быть достаточно данных, полученных в ходе разработки лекарственного препарата и при валидации;
м) распределение частиц по размеру. Условия включения испытания в спецификацию приведены в подпункте «и» пункта 41 настоящих Требований;
н) ресуспендируемость. Условия включения испытания в спецификацию приведены в подпункте «к» пункта 41 настоящих Требований;
о) время восстановления. Условия включения испытания в спецификацию приведены в подпункте «м» пункта 41 настоящих Требований.
Схема решений № 1
Установление критерия приемлемости для контролируемой примеси в активной фармацевтической субстанции
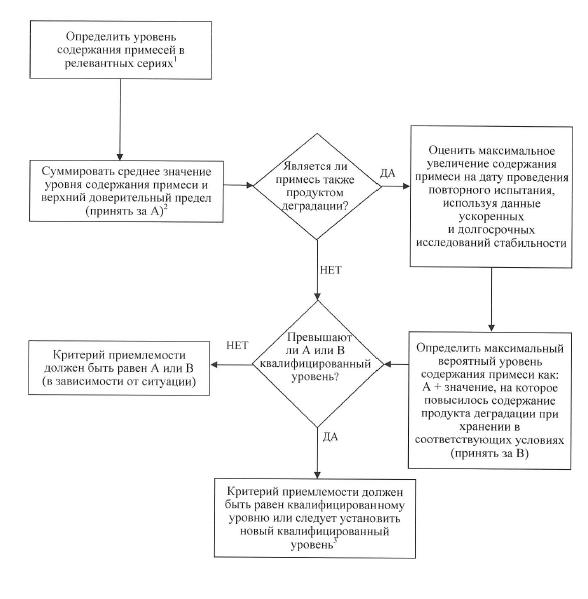
1 Релевантные серии – серии, полученные в ходе исследований на этапах разработки, опытно-промышленного и промышленного производства.
2 Верхний доверительный предел равен стандартному отклонению результатов анализа серий, умноженному на 3.
3 Определяется в соответствии с правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, утверждаемыми Комиссией.
Схема решений № 2
Установление критерия приемлемости для продукта деградации в лекарственном препарате
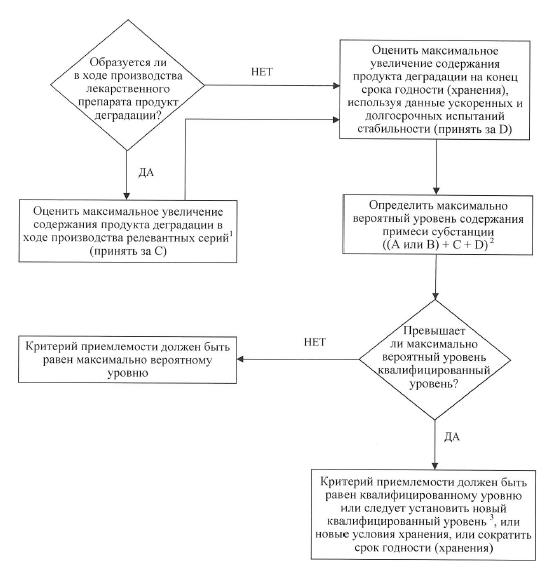
1 Релевантные серии – серии, полученные в ходе исследований на этапах разработки, опытно-промышленного и промышленного производства.
2 Порядок определения A и B представлен в схеме решений № 1.
3 Определяется в соответствии с правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, утверждаемыми Комиссией.
Схема решений № 3
Установление критериев приемлемости относительно размера частиц активной фармацевтической субстанции (АФС)
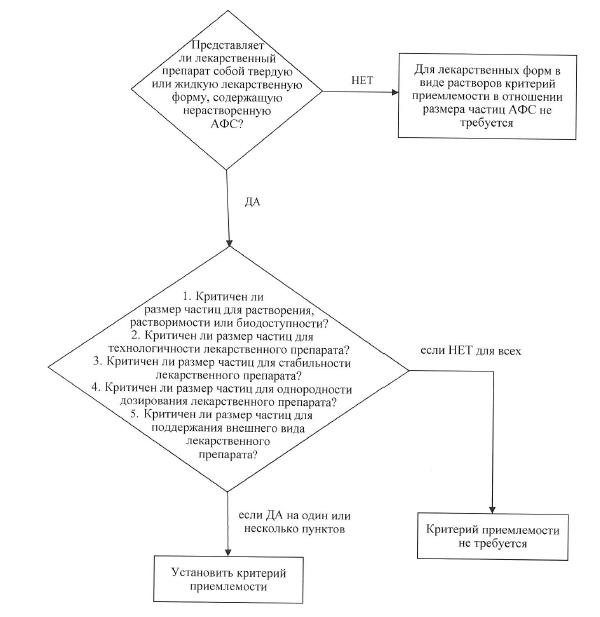
Схема решений № 4
Определение необходимости введения критериев приемлемости относительно полиморфизма активных фармацевтических субстанций (АФС)
и лекарственных препаратов
Активная фармацевтическая субстанция
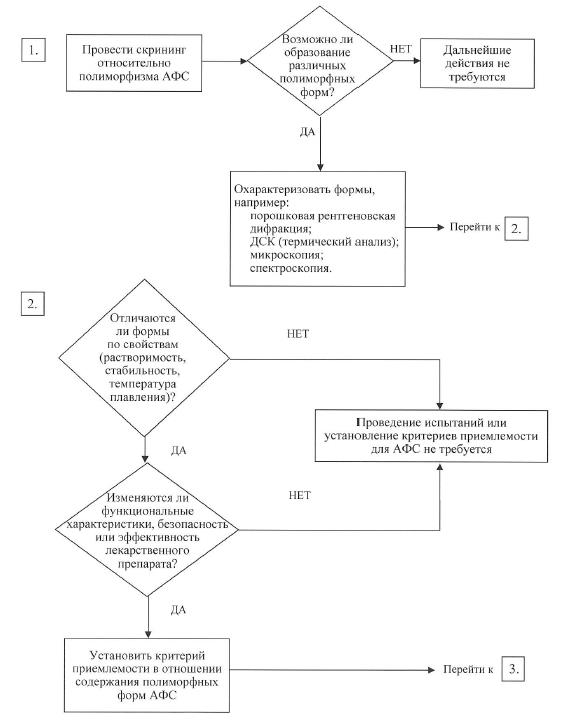
Лекарственный препарат
в виде твердой или жидкой лекарственной формы, содержащей нерастворенную
активную фармацевтическую субстанцию при наличии технической возможности измерения содержания полиморфных форм в лекарственном препарате
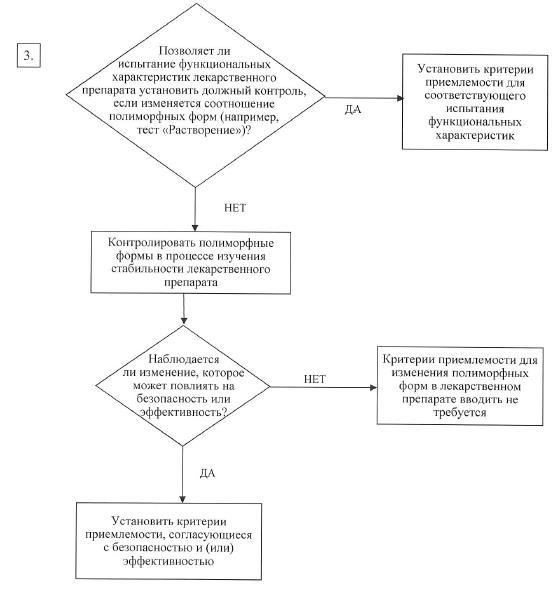
Схема решений № 5
Включение в спецификацию методик: идентификации, количественного определения
и определения энантиомеров-примесей для хиральных активных фармацевтических субстанций (АФС) и лекарственных препаратов, содержащих хиральные активные фармацевтические субстанции
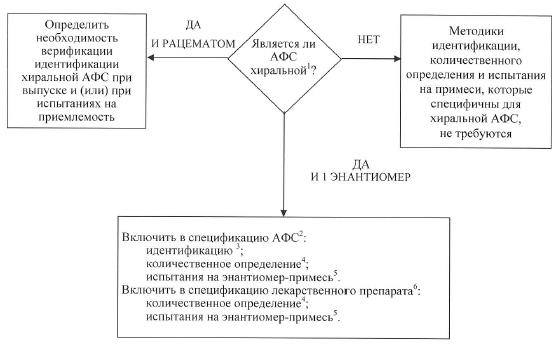
1 Хиральные субстанции природного происхождения в настоящем руководстве не рассматриваются.
2 Как и относительно других примесей, источником которых является исходное сырье, используемое при синтезе активных фармацевтических субстанций, следует предусмотреть контроль качества хиральных веществ, установив пределы для соответствующего исходного сырья или промежуточной продукции, если это обосновано результатами исследований, проведенных в ходе разработки. В основном это случаи, когда имеется несколько хиральных центров (например, 3 и более) или целесообразен контроль на стадии, предшествующей синтезу конечной активной фармацевтической субстанции.
3 Взамен методики идентификации хирального соединения может быть применена методика количественного определения, специфичная для хирального соединения, или методика испытания на энантиомеры-примеси.
4 Взамен методики количественного определения, специфичной для хирального соединения, может быть применена методика количественного определения, неспецифичная относительно хирального соединения, в сочетании с методикой контроля противоположного энантиомера.
5 Содержание противоположного энантиомера в активной фармацевтической субстанции можно определить на основании данных, полученных с помощью методики количественного определения, специфичной относительно хирального соединения, или с помощью отдельной методики.
6 Испытания лекарственного препарата, специфичные относительно стереоизомеров, можно не проводить, если показано, что рацемизация в процессе производства лекарственного препарата и во время хранения готовой лекарственной формы незначительна.
Схема решений № 6
Показатели микробиологической чистоты активных фармацевтических субстанций (АФС) и вспомогательных веществ
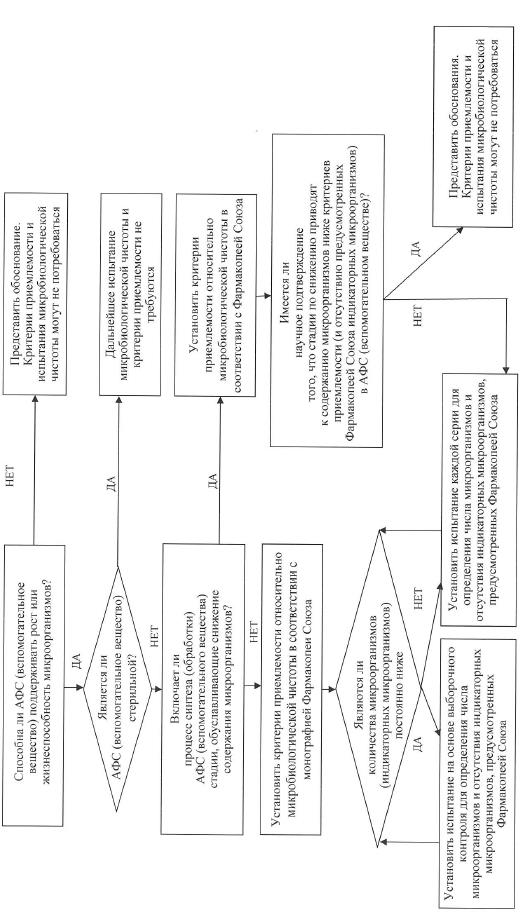
Схема решений № 7
Установление критериев приемлемости для теста «Растворение» лекарственных препаратов
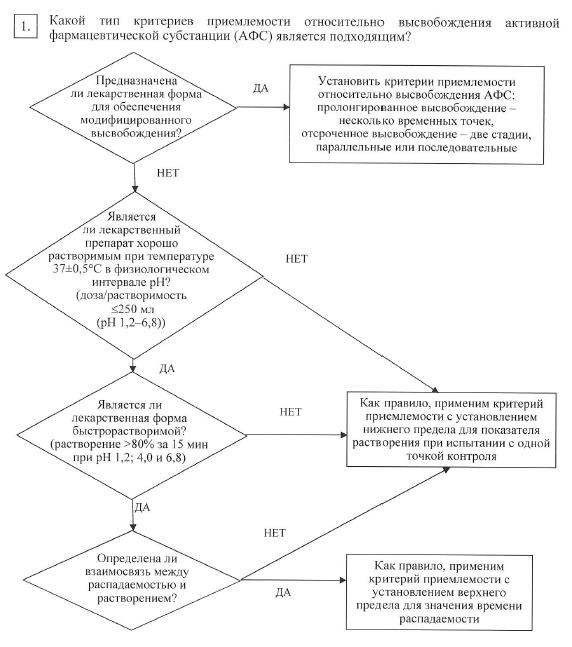
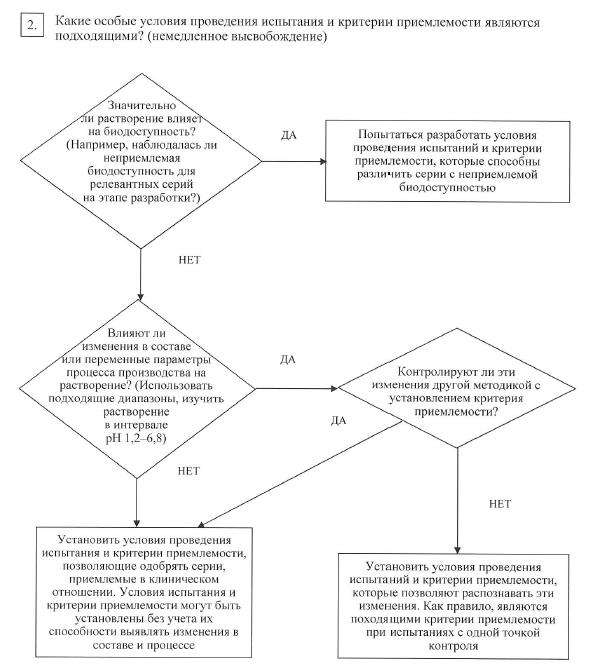
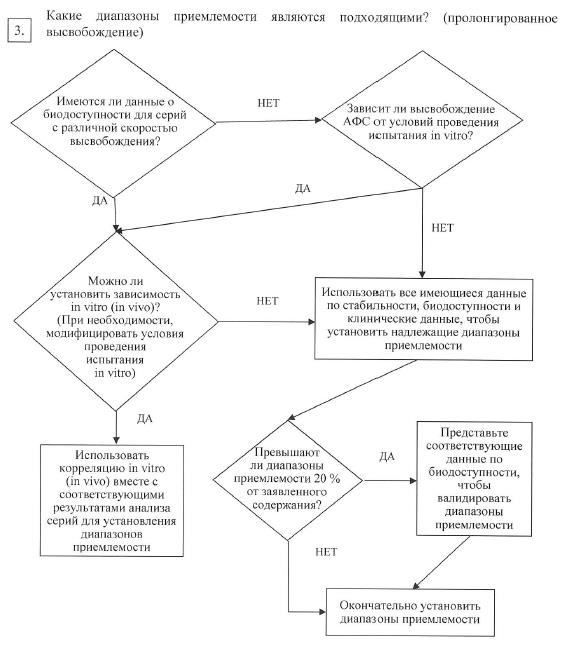
Схема решений № 8
Показатели микробиологической чистоты нестерильных лекарственных препаратов
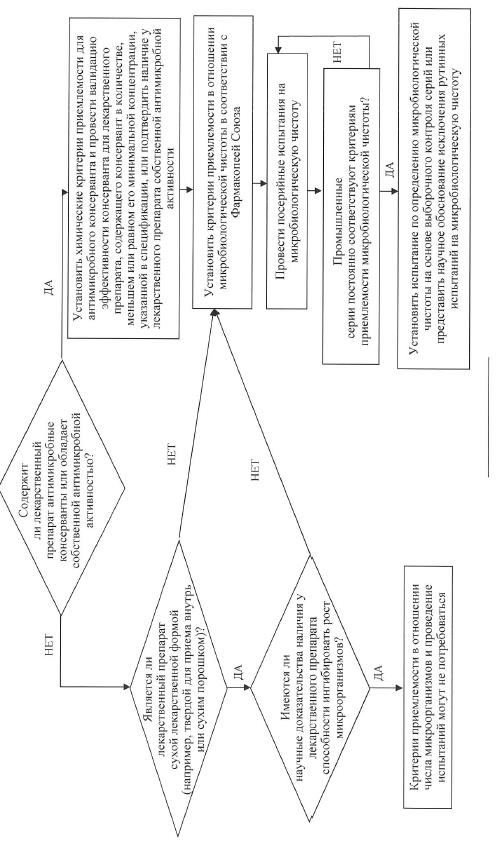
ПРИЛОЖЕНИЕ № 2
к Руководству
по составлению нормативного
документа по качеству
лекарственного препарата
(форма)
ТИТУЛЬНЫЙ ЛИСТ
нормативного документа по качеству
|
СОГЛАСОВАН ____________________________ (наименование уполномоченного органа государства признания) ____________________________ ____________________________ (Ф.И.О., должность, подпись) «____»__________ _____ 20__ г. М.П. |
УТВЕРЖДЕН ____________________________ (наименование уполномоченного органа референтного государства) ____________________________ ____________________________ (Ф.И.О., должность, подпись) «____»_______________ 20__ г. М.П. СОГЛАСОВАН ____________________________ (наименование заявителя или уполномоченного ____________________________ ____________________________ (Ф.И.О., должность, подпись) «____»_______________ 20__ г. М.П. |
НОРМАТИВНЫЙ ДОКУМЕНТ
Торговое наименование лекарственного препарата: ______________________
Международное непатентованное наименование: ________________________
(при его отсутствии – общепринятое (группировочное) наименование, при отсутствии последнего – химическое наименование)
Лекарственная форма: _______________________________________________
Дозировка:_________________________________________________________
Держатель регистрационного удостоверения: __________________________
(наименование и страна держателя регистрационного удостоверения)
Номер и дата нормативного документа: ________________________________
(номер и дата регистрационного удостоверения, выданного референтным государством)
_____________________








